The Reck-Peterson, Villa and Leschziner groups at UC San Diego discovered that the LRRK2 protein, a
mutated subset in Parkinson’s patients, has profound impacts on molecular transport in the cell and
could influence the onset of Parkinson’s disease by playing a role in substantia nigra neuronal death.
The gift of movement is granted to most of us at birth. However, this physical ability can be diminished by numerous pathologies, including the infamous Parkinson’s disease. This particular motor disorder typically manifests during the later stages of one’s life and consists primarily of an inability to elicit intentional movement. Normally, motor control is regulated by dopamine signaling between neurons in two regions of the brain: the basal ganglia and substantia nigra. However, when populations of substantia nigra neurons are destroyed, the basal ganglia neurons no longer receive these essential dopamine signals that permit movement initiation. As a result, patients with Parkinson’s experience hypokinesia, or an inability to initiate and efficiently perform various motor tasks.
Current research on Parkinson’s revolves around understanding the cause of substantia nigra neuron death. Researchers are currently examining the precise mechanics of neuronal death, as well as the role of supportive brain cells, known as glial cells, in maintaining the health of substantia nigra neurons. However, a novel and fascinating approach to elucidating Parkinson’s explores the role of molecular transport in neuronal decay and may bring insight to future treatments for this devastating disease.
Molecular transport is vital for proper cellular function in all cell types. Materials must be delivered to certain locations in the cell in a timely manner since organelles and vesicles cannot passively move to the target cellular location. For this reason, vesicular or organellar cargoes are linked to molecular motors that physically travel along long filament polymers known as microtubules, which are commonly referred to as cellular highways. Molecular motors traffic their cargoes to target locations in the cell by traveling along these highways.
In neurons, numerous processes require molecular transport. For example, synaptic neurotransmitter release requires the trafficking of vesicles containing the neurotransmitter of interest to the axon terminal. Mitochondria are trafficked throughout neurons to ensure that the energy levels required to sustain neuronal function are met. Endoplasmic reticula act as crucial calcium stores in neurons and must be localized to areas that require calcium for signaling processes. Finally, molecular transport systems remove cellular toxins and damage in cells, so impeding molecular transport could prove lethal to neurons.
Molecular transport systems are especially important in neurons due to their elongated morphology. Materials are constantly shuttled between the neuronal cell body, or the soma, and axon terminals. The two types of molecular motors involved in the transport system are kinesin and dynein. Generally, kinesins move materials towards the negative end of the microtubule, from the soma to the axon terminals, while dyneins function in the reverse. Clearly, a lack of coordinated molecular transport impedes neuronal function and health.
A particular protein known as the Leucine Rich Repeat Kinase 2 (LRRK2) has been found to influence molecular transport and is mutated in a subset of patients with Parkinson’s disease. The mysterious LRRK2 belongs to a group of enzymes known as kinases, which initiate cellular transduction signals by phosphorylating amino acids on other cellular proteins. Phosphorylation of proteins can cause a wide variety of effects, including changing protein-protein interactions. While LRRK2’s normal function in a healthy cell is not yet known, the kinase is known to oligomerize, or form long protein chains by binding to other identical LRRK2 kinases. These LRRK2 oligomers can then wrap around microtubules in a helical manner.¹ Like the dual-faced Roman deity Janus, personified to preside over duality and transitions, LRRK2 function remains mysterious in both healthy and diseased cells.
Dr. Samara Reck-Peterson, Professor of Cellular and Molecular Biology and the Division of Biological Sciences at UC San Diego and a Howard Hughes Medical Institute Investigator, is currently exploring wildtype and mutant LRRK2’s roles in cellular function and Parkinson’s disease respectively. The investigation of LRRK2 has been an ongoing project between the Reck-Peterson group and several other UC San Diego research laboratories. LRRK2 oligomers were first observed by the laboratory of Dr. Mark Ellisman, and later, the laboratories of Dr. Andres Leschziner and Dr. Elizabeth Villa solved the three-dimensional structures of single LRRK2 subunits¹ and LRRK2 oligomers on microtubules respectively.² Using electron microscopy, microtubule-associated oligomers were resolved in the native cell environment and a high-resolution structure of LRRK2 in isolation was determined as well.
The LRRK2 kinase exists in two conformations: closed and open. In its closed conformation, the kinase is active and can phosphorylate its molecular targets. By comparing the structures of the microtubule-associated LRRK2 oligomers with LRRK2 in isolation, Drs. Leschziner and Villa’s group discovered that the kinase domain was closed in the long linear oligomer chains that wrap around microtubules in a helical manner,¹² while Dr. Reck-Peterson’s group discovered that these oligomeric chains form obstructions that can block molecular motor movement along a microtubule.¹ It is hypothesized that these oligomeric chains can perhaps obstruct molecular motors from delivering their cargoes to their target site in the cell, jeopardizing cellular health.

Two conformations of LRRK2 protein monomer. Upper: LRRK2 in open and inactive conformation. Lower: LRRK2 in closed and active conformation.
Other molecules linked to neurodegenerative diseases, such as Tau or MAPs, can also generate roadblocks for molecular motors. While Tau and some MAPs latch onto one small portion of the microtubule, LRRK2 oligomers wrap around the microtubule in a helical fashion, making it harder for molecular motors to step past them.¹
LRRK2 oligomers are also unique in that they obstruct the movement of both dynein and kinesin. While kinesin and dynein motors both transport cargoes along the same microtubular highways, their structural differences allow each motor to step along microtubules and navigate microtubular roadblocks in distinct ways. Microtubules themselves usually consist of thirteen proto-filament chains interconnected together to form a hollow, tubular structure. Each proto-filament is built from dimers of two subunits, alpha tubulin and beta tubulin.
Kinesin and dynein have different patterns of stepping along microtubules. Kinesin is composed of two identical motor heads connected by short linker chains to the central stalk of the motor complex. The kinesin motor heads take small alternating steps along a single microtubule protofilament, stepping from one tubulin dimer to the next. In contrast, the dynein motor is much larger, can take steps of varying sizes, and is not restricted to moving along a single protofilament. This greater flexibility in dynein’s movement allows it to sidestep small and localized microtubular obstructions, which kinesin is unable to do.
LRRK2 oligomers are large enough to span multiple microtubule protofilaments, a fact that perhaps explains why it can so potently inhibit both dynein and kinesin. While normal LRRK2 can also form microtubular oligomers, four mutations in LRRK2 that are linked to Parkinson’s disease correlate with the oligomer formation becoming more probable.¹ The exact mechanism of how these mutations impact LRRK2’s ability to oligomerize is not yet known, though it is hypothesized that the mutated kinase may perhaps spend more time in its closed conformation. A growing theory on the cause of Parkinson’s disease concerns microglia, or macrophage-like glial cells that work to remove and repair damaged parts of a neuron. These microglia cells release chemical signals into the extracellular fluid to accelerate neuronal repair mechanisms. However, in order for the signals to take effect, molecular motors are required to transport these signals within the microglia to the release terminals upon appropriate stimulation. If molecular transport is inhibited in the microglia that support adjacent substantia nigra neurons, damage could accumulate in these nigral cells and lead to their programmed death. Thus, some researchers apart from the Reck-Peterson group have begun to postulate that rather than a direct malfunction in the nigral neurons, microglial dysfunction may instead lead to the substantia nigra neuron death that is characteristic of Parkinson’s disease.
LRRK2’s role as a kinase is also being actively researched. Recently, Rab GTPases have been identified as LRRK2 phosphorylation targets.¹ There are about sixty types of Rab GTPases known in humans, and their unifying function is to signal for the fusion of lipid bilayer membranes. For example, Rab GTPases ensure the fusion of membranes during vesicular exocytosis, a process by which chemical messengers are released from a cell into the extracellular environment. Rab GTPases also mediate the linkage between a cargo-filled vesicle and molecular motor. In order to link a cargo to a motor, adaptor proteins are used to connect vesicle-linked Rabs to motors. One hypothesis currently being examined is whether the LRRK2 phosphorylation of Rabs alters the ability of the motors to link to their cargoes. An examination of this hypothesis will reveal whether LRRK2 impacts motor-directed cargo transport in ways other than the microtubular oligomer roadblocks in the cell, perhaps lending another clue to the kinase’s role in the onset of Parkinson’s disease.
Kinases such as LRRK2 are excellent drug targets for possible Parkinson’s disease therapeutics. Two groups of kinase inhibitors, known as the Type I and Type II inhibitors, have been explored in relation to LRRK2. Generally, Type I inhibitors lock kinases in their closed, active conformation, while Type II inhibitors lock kinases in their open, inactive conformation. The inhibitors accomplish this by binding to the ATP-binding pocket at several conserved motifs either in the closed or open kinase, locking the kinase in the given conformation.³
Kinases are active in their closed conformation and inactive in their open conformation. Therefore, it was hypothesized that Type II inhibitors might rescue molecular motor movement on microtubules by keeping the kinase in its open and inactive conformation, inhibiting its ability to oligomerize. Dr. Reck-Peterson’s lab directly tested this hypothesis by using pure proteins in vitro, and it was found that LRRK2 treated with Type II inhibitors no longer acted as a microtubular roadblock for kinesin and dynein motors. In contrast, Type I inhibitors caused the kinesin motor to be blocked at even higher levels by LRRK2 oligomers. The Reck-Peterson group also found that in eukaryotic cell culture, Type II inhibitors decrease LRRK2’s ability to form oligomer filaments around microtubules.¹
As the Reck-Peterson, Leschziner and Villa groups at UC San Diego look into the future of LRRK2 and Parkinson’s research, many questions remain to be answered regarding the exact role of LRRK2 in Parkinson’s development. The three labs, along with two collaborating labs in Germany, have recently received over seven million dollars in funding from Aligning Science Across Parkinson’s Disease to explore this question. The groups plan to determine how both wild type and mutant LRRK2 impact intracellular trafficking, how the mutant may lead to Parkinson’s disease, and how wild type LRRK2 functions in normal cells. It remains to be determined whether the mutated forms of LRRK2 disrupt the substantia nigra neurons by jeopardizing the health of the nigral neurons themselves, or the health of the supporting microglia. By understanding the role of the mysterious and two-faced LRRK2 in substantia nigra neuronal death, researchers will come yet another step closer to solving the puzzling mystery of Parkinson’s disease and restoring patients’ ability to efficiently move.
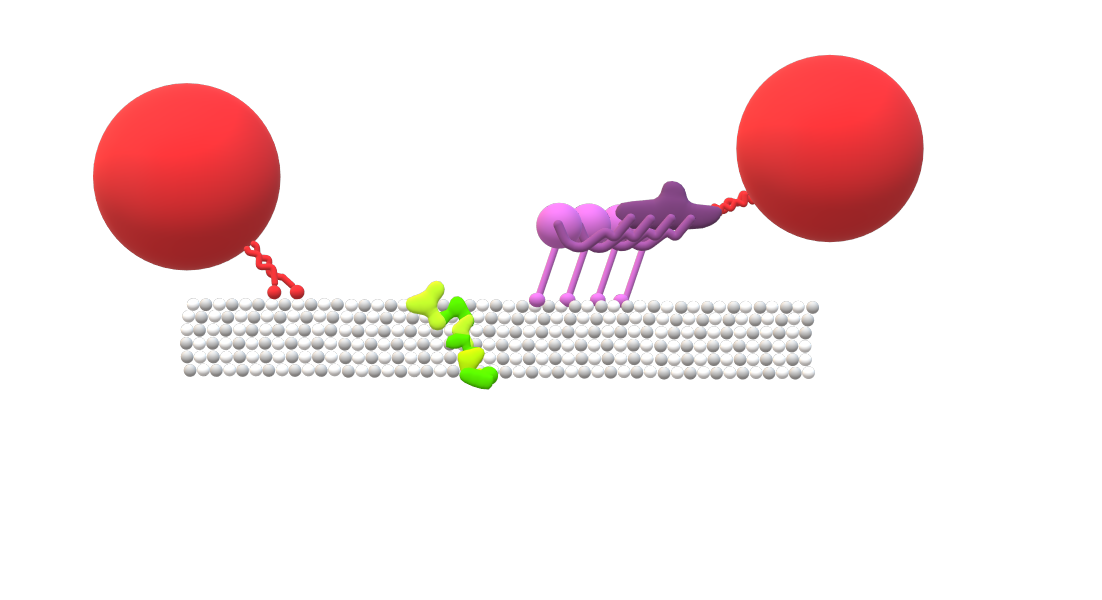
Six microtubule protofilaments. LRRK2 oligomer (green) obstructs the movement of both dynein (purple) towards the positive end of the microtubule, and kinesin (red) towards the negative end. Kinesin motor heads are connected by short linkers to the central stalk of the motor complex, while the dynein motor domains exhibit larger movement flexibility.
REFERENCES
- Deniston CK, Salogiannis J, Mathea S, Snead DM, Lahiri I, Matyszewski M, Donosa O, Watanabe R, Böhning J, Shiau AK, Knapp S, Villa E, Reck-Peterson SL, Leschziner AE. 2020. Structure of LRRK2 in Parkinson’s disease and model for microtubule interaction. Nature. 588(7837):344-349. doi: 10.1038/s41586-020-2673-2. Epub 2020 Aug 19. PMID: 32814344; PMCID: PMC7726071. https://pubmed.ncbi.nlm.nih.gov/32814344/
- Watanabe R, Buschauer R, Böhning J, Audagnotto M, Lasker K, Lu TW, Boassa D, Taylor S, Villa E. 2020. The In Situ Structure of Parkinson’s Disease-Linked LRRK2. Cell. 2020 Sep 17;182(6):1508-1518.e16. doi: 10.1016/j.cell.2020.08.004. Epub. PMID: 32783917; PMCID: PMC7869717. https://pubmed.ncbi.nlm.nih.gov/32783917/
- Roskoski R. 2015. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Elsevier. http://www.brimr.org/Reprints/149.pdf
WRITTEN BY ANNA HAKIMI
Anna is a Neurobiology Major from Roger Revelle College. She will be graduating in 2022.
FROM SALTMAN QUARTERLY VOL. 18
To read the original version, please click here. To read the full version on our website, please click here. To read more individual articles, please click here.