,_H%26E.jpg){kind=link}
In 1732, a shepherd in Spain noticed a sheep compulsively scraping against a fence, a behavior that soon spread throughout the flock and left every sheep deceased. The infectious disease causing this symptom became known as “scrapie,” and its cause remained unknown for many years. Today, this condition is classified as a transmissible spongiform encephalopathy, a category of diseases which includes Bovine Spongiform Encephalopathy, or “Mad Cow Disease,” in cattle, and Creutzfeldt-Jakob Disease (CJD) in humans. Prion proteins were identified as the cause of these infectious neurodegenerative diseases, which became known as prion protein diseases, and are a puzzling subject to molecular biologists.
Prion proteins originate as normal, functional proteins in all mammals; the highly conserved gene for these proteins, named PRNP, is found in every mammalian genome. Like any other protein, prion proteins are formed through protein folding mechanisms, which involve transforming amino acid chains into three-dimensional protein structures and thus determine proper protein function. Although numerous checkpoints ensure proper folding, misfolding can still occur due to various genetic and environmental stimuli, causing disease in the organism. Therefore, prion proteins are misfolded, disease-causing proteins localized to the brain as a result of errors in protein folding mechanisms.
While diseases of bacterial or viral nature replicate via genomically driven mechanisms, prion proteins have no nucleic genome. This allows prion protein diseases to be resistant to forms of elimination that target nucleic acid compounds, such as UV light radiation. Instead, prion proteins spread by binding to similar proteins to form insoluble proteinaceous aggregates, or masses. When an infectious prion protein with abnormal structure (PrPSc) binds to a normal cellular prion protein (PrPC), it converts the PrPC into a PrPSc, causing it to acquire the diseased phenotype. The newly made infectious PrPSc continues this process, resulting in mass amounts of PrPSc that clump into aggregates. The details of this replication are poorly understood, and a greater understanding of how this polymerization occurs will be necessary to make advances against prion protein diseases.
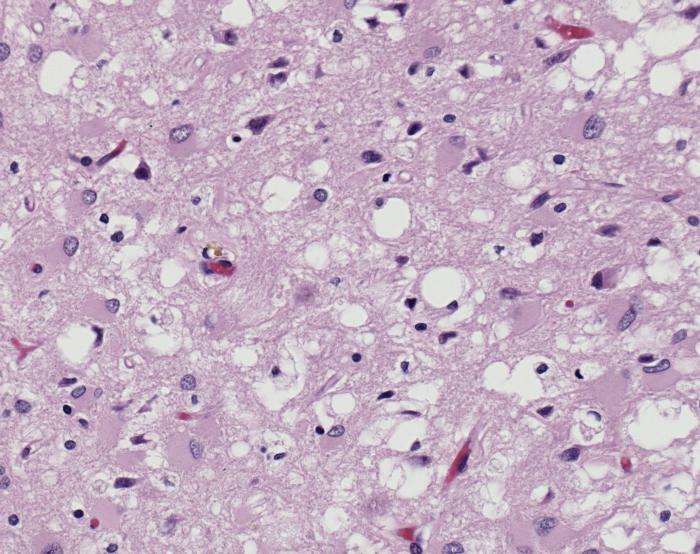
One of the major differences between normal PrPC and infectious PrPSc is their resistance to degradation. PrPC is deteriorated by enzymes called proteases that regulate protein activity through the hydrolysis of peptide bonds, or the breaking of linkages between amino acids that constitute proteins. On the contrary, PrPSc is resistant to protease degradation, and instead clumps together in insoluble protein aggregates, causing vacuoles, or holes, in the brain. These vacuoles can be seen in all prion protein diseases, and result in neuron loss and subsequent neurodegeneration.
One prion protein disease that has garnered public attention is Bovine Spongiform Encephalopathy (BSE). BSE, or “Mad Cow Disease,” causes the symptomatic loss of central nervous control and consequent death. Hypothesized to have originated from the cross-contamination of “scrapie” sheep meat with live cattle in the United Kingdom, BSE became an epidemic in the late 1980s and 1990s and catalyzed a greater push for research on prion proteins. Between 1986 and 1993, approximately 120,000 cattle were diagnosed with BSE in Britain, but as of 2017, there are no known novel cases. This epidemic promoted the development of major disease prevention strategies worldwide, including the United States restricting European imports of ruminant mammals and banning ruminant feeds containing animal proteins and tissue. These restriction techniques exist to prevent transmission to humans who are capable of contracting prion protein diseases as well.
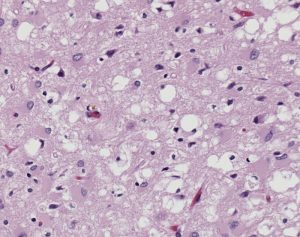
The major prion protein disease that infects humans is Creutzfeldt-Jakob Disease (CJD). It is approximated that one person per million is affected by CJD internationally every year, resulting in about three hundred cases per year in the United States. The symptoms of CJD include cognitive deterioration, stiff muscles, and ataxia, or the loss of bodily control over day to day movements such as walking and swallowing. Like all prion protein diseases, CJD is fatal. Combined with its highly infectious nature, the fatality of CJD pushes research efforts on how this deadly disease is transmitted.
CJD can be acquired in three ways. About ninety percent of cases in the United States are acquired sporadically, meaning the disease occurs as a random event in the body. In sporadic cases, a normal PrPC undergoes a random change in protein folding that converts it into an infectious PrPSc, causing the disease to spread exponentially via the self-replicating polymerization of diseased PrPSc aggregates. Although sporadic cases are not necessarily linked to genetic mutations, the connection is still possible and is being actively investigated by researchers.
In contrast, the second form of CJD is acquired genetically and accounts for about ten percent of cases in the United States. This form of CJD originates from a mutation in an autosomal dominant trait, or a trait whose gene is on a non-sex chromosome. Specifically, CJD is genetically induced by multiple mutations on the autosomal dominant PRNP gene that increase the likelihood of acquiring PrPSc and subsequently CJD.
CJD can also be acquired through the direct transmission of the disease, which is attributed to less than one percent of cases in the United States. This transmission occurs through the consumption of meat infected by BSE, or by medical equipment contaminated with CJD or another prion protein disease. When acquired through transmission from BSE, the disease is known as variant Creutzfeldt-Jakob Disease (vCJD). Concerns over vCJD from infected meat consumption became a mainstream concern during the BSE epidemic of the 1990s, yet remain of lesser concern today.
From their relatively recent identification, unique replication processes, and fatal outcomes, prion protein diseases differ from many other well-known diseases. With the recent epidemic of BSE in cattle and danger of possible spread to humans, prions remain an important topic for biologists to investigate. Continual research about how prion proteins function, and in particular how these misfolded proteins infect and degrade the brain, is necessary to overcome these gaps of knowledge and challenge the spread of these fatal diseases.
References:
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4626585/
- https://www.accessscience.com/content/prion/545350
- https://www.cdc.gov/prions/bse/prevention.html
- https://medlineplus.gov/genetics/gene/prnp/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2576539/
- https://www.accessscience.com/content/prion-disease/757460
- https://pubmed.ncbi.nlm.nih.gov/29887132/
- https://cjdfoundation.org/types-prion-diseases
- https://www.sciencedirect.com/topics/nursing-and-health-professions/ataxia
- https://www.accessscience.com/content/protein-folding/801070
- https://www.accessscience.com/content/human-genetics/324600
- https://www.centerforfoodsafety.org/issues/1040/mad-cow-disease/timeline-mad-cow-disease-outbreaks