

Consider this: you have been working in a lab for a few years, and you think that you have discovered a new drug to combat adolescent depression. Now, you have to test the efficacy and safety of the drug in a series of clinical trials in order to obtain FDA approval for it to go on the market. Of course, most people know that getting FDA approval for a drug requires several stringent guidelines to be met, and that meeting these guidelines is no easy task. However, this was not always the case. Before the 1960s, the FDA existed in name, but its regulatory functions were lax at best, and negligent at worst. The turning point occurred in the early 1960s after the thalidomide tragedy, a disaster which demonstrates the importance of regulating the pharmaceutical industry.
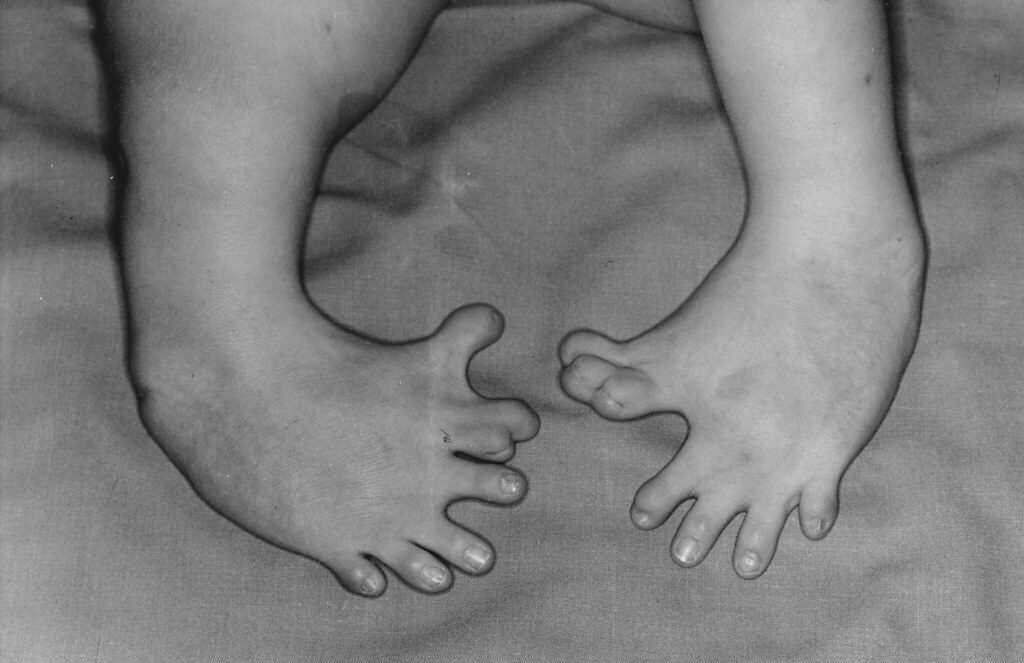
The thalidomide tragedy occurred in the 1950s, a time when the standards of regulation in the pharmaceutical industry were not nearly as rigorous as they are today. Thalidomide was first marketed in Germany during the mid-1950s as a drug to aid with sleeping problems and insomnia. The drug’s safety was emphasized heavily, and it was advertised as being acceptable for anyone to ingest, including pregnant women and children. In fact, the drug’s developers confidently proclaimed that they “‘could not find a dose [of thalidomide] high enough to kill a rat.’” Thalidomide sales skyrocketed, and by 1960, the drug was marketed in 46 countries. At this time, research on thalidomide had begun to show the drug’s effectiveness in alleviating nausea in pregnant women, and many physicians started prescribing the drug off-label as a treatment for morning sickness. Not long after thalidomide started being used for this purpose, scientists began to observe birth defects in children born to mothers who had taken thalidomide during their pregnancy; studies showed that exposure was particularly dangerous for infants born to mothers that had used the drug approximately 20—34 days post-fertilization. Common birth defects seen in these children included deletion of the ears, deafness, severe underdevelopment or absence of the arms, defects in the femur and tibia (bones of the legs), and many more. More than 10,000 children around the world were born with thalidomide-related birth defects, and tragically, many of these children, now adults, still suffer from the effects of thalidomide.
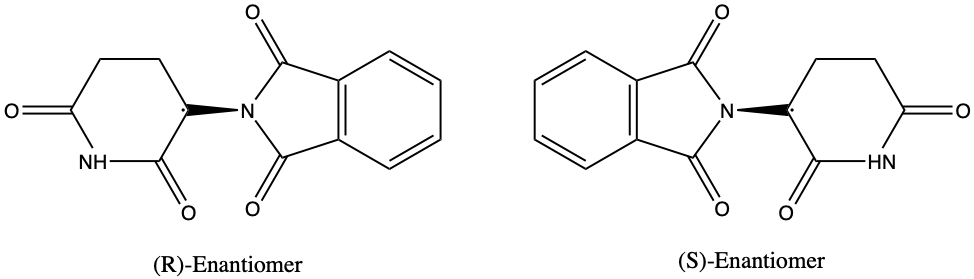
What exactly is the chemistry behind this tragedy? This is where the concept of enantiomers comes in. The thalidomide molecule occurs naturally in two forms, (R)-thalidomide and (S)- thalidomide, which are enantiomers, or mirror image molecules, of each other. The (R)-form has remedial properties, while the (S)-form is a teratogen, an agent that can cause birth defects. However, in the body, the two enantiomers of thalidomide constantly interconvert to produce a mixture in which both enantiomers of the drug are present, making it extremely difficult and risky to isolate purely (R)-thalidomide. Unfortunately, the stereochemistry of thalidomide and its profound effect on the human body was not realized in the 1950s and 1960s when the drug was prescribed to many pregnant women.
Although the thalidomide disaster had a widespread impact in Europe, thalidomide’s effects were not as prevalent in the United States. Why? In 1961, as the drug became popular in Germany and other European nations, an application was submitted to the FDA to gain approval for marketing thalidomide in the United States. However, Frances Oldham Kelsey, a new FDA employee and physician, was wary of approving the drug, for she had seen studies published in Germany showing an association between thalidomide usage and peripheral neuritis (general problems of the peripheral nervous system). Kelsey insisted on getting more information about the drug before approving it as a remedy for morning sickness; she was particularly concerned about whether thalidomide would be able to cross the placenta in pregnant mothers-to-be, potentially harming their unborn children. Dr. Kelsey’s hesitations about thalidomide proved to be correct–towards the end of 1961, studies began to be published which detailed the drug’s harmful effects during gestation. Unfortunately, thalidomide had been distributed to around 20,000 patients nationwide while in its clinical trial phase; at least 207 of these patients were pregnant women, and 17 of these women reported birth defects in their children. Had Dr. Kelsey not hesitated to approve the drug, this figure would have likely been much higher.
The thalidomide tragedy served as a brutal wake-up call for the United States Food and Drug Administration (FDA) on their policies regarding clinical trials and pharmaceutical regulations. Following the thalidomide incident, the Kefauver-Harris Amendments were passed in 1962, requiring “that for any sponsor of a drug, usually a company, that planned to investigate that drug clinically, that sponsor had to provide the FDA with a detailed outline of the study.” Additionally, the sponsors and researchers would be responsible for keeping meticulous records of the findings and periodically reporting them to the FDA. This provided a stark contrast to the previous lax guidelines of the FDA. For example, Dr. Frances Oldham Kelsey described in her memoir drug industry practices in the 1960s that would be considered completely abhorrent today. For example, drugs had a sixty day review period; if a drug was neither approved nor rejected in sixty days or less, it would automatically be put on the market. Following the thalidomide event, regulations were made stricter than this. Nevertheless, the FDA still continues to be peppered with issues.
One of the most serious problems regarding the FDA, particularly in recent years, has been its tendency to release serious warnings about drugs after they have been placed on the market. With mounting governmental pressures to accelerate the drug approval process, 71 out of 222 drugs placed on the market between 2001 and 2010 came with side effects so serious that the FDA issued black-box warnings (warnings pertaining to potentially serious or lethal side effects of a prescription drug) for these treatments. What is even more concerning is that these safety concerns only came to light, on average, four years after the drugs’ market approval. Common drugs with black-box warnings include Abilify (generic name aripiprazole; used to treat depression but can cause suicidal ideation in youth), Accutane (generic name isotretinoin; used to combat acne in youth but can lead to birth defects), and Cipro (generic name ciprofloxacin; an antibiotic which has been implicated with tendon rupture).
Overall, the thalidomide disaster is one that still remains fresh in the lives of those affected by it. The result of a combination of poor drug regulation and incomplete knowledge of the drug’s chemical properties, this tragedy serves as a reminder of the importance of thorough research and regulation before the approval of any drug. Though important changes were implemented by the FDA after this disaster, the agency still continues to face problems regarding the approval of pharmaceuticals, and there is much room for improvement in this area.
Sources:
- https://www.britannica.com/science/thalidomide
- https://helix.northwestern.edu/article/thalidomide-tragedy-lessons-drug-safety-and-regulation
- https://embryo.asu.edu/pages/us-regulatory-response-thalidomide-1950-2000
- https://academic.oup.com/toxsci/article/122/1/1/1672454
- https://thalidomide.ca/en/congenital-malformations/
- http://broughttolife.sciencemuseum.org.uk/broughttolife/themes/controversies/thalidomide
- https://www.acs.org/content/acs/en/molecule-of-the-week/archive/t/thalidomide.html
- https://chw.org/medical-care/genetics-and-genomics-program/medical-genetics/teratogens
- https://www.science.oregonstate.edu/~gablek/CH334/Chapter5/Thalidomide.htm
- https://www.nytimes.com/2015/08/08/science/frances-oldham-kelsey-fda-doctor-who-exposed-danger-of-thalidomide-dies-at-101.html
- https://www.fda.gov/regulatory-information/laws-enforced-fda
- https://www.smithsonianmag.com/science-nature/woman-who-stood-between-america-and-epidemic-birth-defects-180963165/
- https://www.npr.org/sections/health-shots/2017/05/09/527575055/one-third-of-new-drugs-had-safety-problems-after-fda-approval
- https://www.drugwatch.com/fda/black-box-warnings/
