
ABSTRACT
Rheumatoid arthritis (RA) is a debilitating autoimmune disease that causes painful inflammation within joint tissue. Despite the success of modern therapeutics in treating detectable inflammation, patients with RA often experience lingering neuropathic pain. Neuropathic pain is a chronic burning sensation that results from a disruption of the somatosensory nervous system and extensive nerve damage. This study focuses on identifying potential connections between neuropathic pain and the inflammatory response that is characteristic of RA. These connections were determined and analyzed using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) program application. Genes previously identified as relating to human disease in both mouse models and human genes were first entered into STRING. Based on the quantity and strength of the connections generated, several genes were identified as potential critical intersection points for the development of neuropathic pain. The assessment included several characteristics of the genes such as physical proximity, pathway relation, co-expression, and their history of co-mention in previous PubMed abstracts. The genes identified in this study as critical intersection points for neuropathic pain development can be further studied to develop possible therapeutics against RA.
INTRODUCTION
Rheumatoid arthritis (RA) is an autoimmune disease where the immune system causes joint inflammation, potentially resulting in permanent damage and deformity.¹ This form of arthritis affects women three times more often than men, and in severe cases, it disrupts organ functionality.¹ An estimated 1.3 million people, making up one percent of the world’s population, are afflicted with this disease.¹ Common symptoms of RA include joint swelling and tactile pain on the limbs. The chronic pain characteristic of RA does not necessarily correlate with detected levels of joint swelling. There is a range in the levels of joint swelling in relation to pain that patients experience. Specifically, the onset of pain prior to swelling is a main complaint from RA patients.²
The pain induced by RA is often described as gnawing or aching, descriptions which have often been associated with nociceptive pain caused by tissue damage. Joint structures are lined with nociceptive neurons, whose cell bodies are located in the dorsal root ganglion. During the inflammatory process, immune cells release proinflammatory cytokines while nociceptive cells release calcitonin gene-related peptides which sensitize the primary afferent neuron, which relays the pain signal to the CNS. As a result, patients experience amplified pain in the joints and spinal column. This mechanism outlines the onset and effects of nociceptive pain.⁴
In contrast to this nociceptive pain, many RA patients also use descriptors that are typical of neuropathic pain: the perception of pain in the absence of tissue damage or nociceptive input. Neuropathic pain is usually caused by damage to central and peripheral nerves and causes sensations such as burning or prickling.⁴ Unlike nociceptive pain, the mechanism for neuropathic pain is not yet completely understood; however, it is assumed to be caused by a lesion or disease affecting the somatosensory nervous system, thus leading to the perception of pain even in the absence of pain stimuli. It is maladaptive (impairs daily motor functions) and persists with minimal or undetected peripheral inflammatory pathology. Patients are diagnosed with neuropathic pain using a combination of sensory tests and clinical expertise.⁵ Symptoms include several abnormal sensations such as allodynia, or pain caused by a stimulus that does not induce pain under normal conditions. The neuropathic element of pain in chronic RA has been previously well documented; therefore, this project aims to investigate the role of the neuropathic element of pain in the larger scheme of an inflammatory response.
Specific antibodies target the synovium (soft tissue that lines joint cavities) and cartilage, leading to the progression of RA. Recent studies show that epigenetic disorders such as abnormal histone modification and DNA methylation contribute to the generation of rheumatoid arthritis synovial fibroblasts (RASF).³ RASF lines the joint and secrete degrading enzymes, proinflammatory cytokines, and chemokines that generate inflammation.² This inflammation activates osteoclasts, or cells that break down bone tissue, which can cause permanent bone damage.
Current treatment options for RA include non-steroidal anti-inflammatory drugs which block cyclooxygenase, an enzyme which is used to make prostaglandins. Prostaglandins respond to injury in the body and cause inflammation. Consequently, blocking cyclooxygenase relieves inflammation. Another treatment option includes disease-modifying antirheumatic drugs. This class of drugs works to treat RA symptoms by suppressing the body’s immune and inflammatory responses. The mechanisms of these drugs often operate by inhibiting the transcription of specific genes in inflammatory and immune pathways. The wide range of RA symptoms makes it challenging to discover therapeutics that address both pain and swelling. Although several effective anti-inflammatory treatments have been developed, current therapies targeted at effectively treating inflammation inadequately address the accompanying chronic pain patients’ experiences.
METHODOLOGY
In this project we aimed to investigate the role of the neuropathic element of pain in the larger scheme of an inflammatory response. A literature search was conducted utilizing the PubMed search engine for known genes associated with inflammation, neuropathic pain, nociceptive pain, and proteins in these pathways. The following terms were used: inflammation, neuropathic pain, immune response, and nociceptive pain. The immune pathways are incredibly complex and consist of many cellular messenger cascades including cytokine mediators, phosphorylation complexes, and a variety of cross-genomic interactions. Due to this inherent complexity, the literature provided an abundance of relevant genes. These genes included promoters, as well as growth factors, transcription factors, cytokines, and a variety of other ligands. All of the ligands we found work together in the cell signaling of nociceptive pain, neuropathic pain, inflammation, and immune response. A collection of 150 genes were compiled from multiple articles and then entered into STRING, an open access biological database and web-based resource of known and predicted interactions between proteins (https://string-db.org/). The program provided a plethora of information including background information on each protein, co-expression rates, relevant pathways, and tissue expression. STRING then ranked the most highly connected genes. From this initial screening, candidate genes that were observed to have the highest densities of correlations with other genes were selected and re-entered into STRING for further analysis. More specific analysis of the relationship between these genes may assist in identifying the larger role these genes may play in the pathology of RA, and more specifically neuropathic pain, as the pathologies of these genes overlap with those proven to be drivers of RA and neuropathic pain.
RESULTS
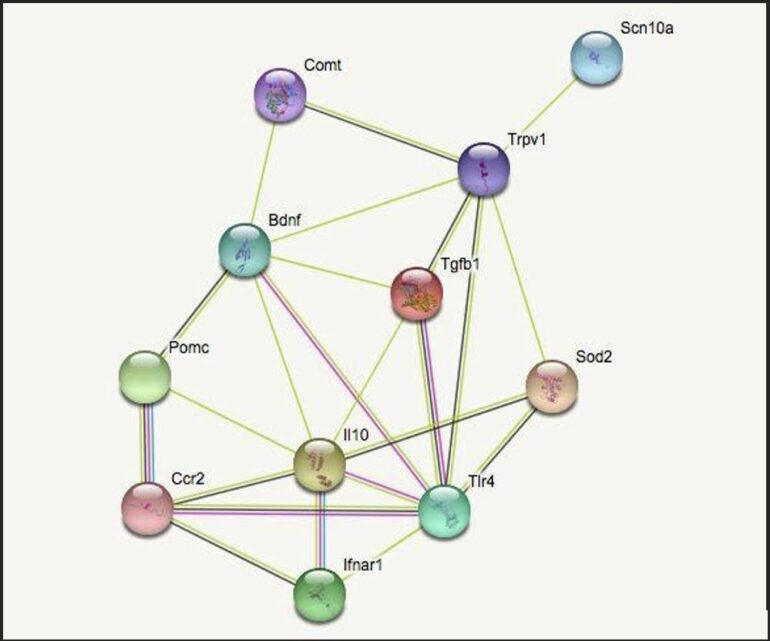
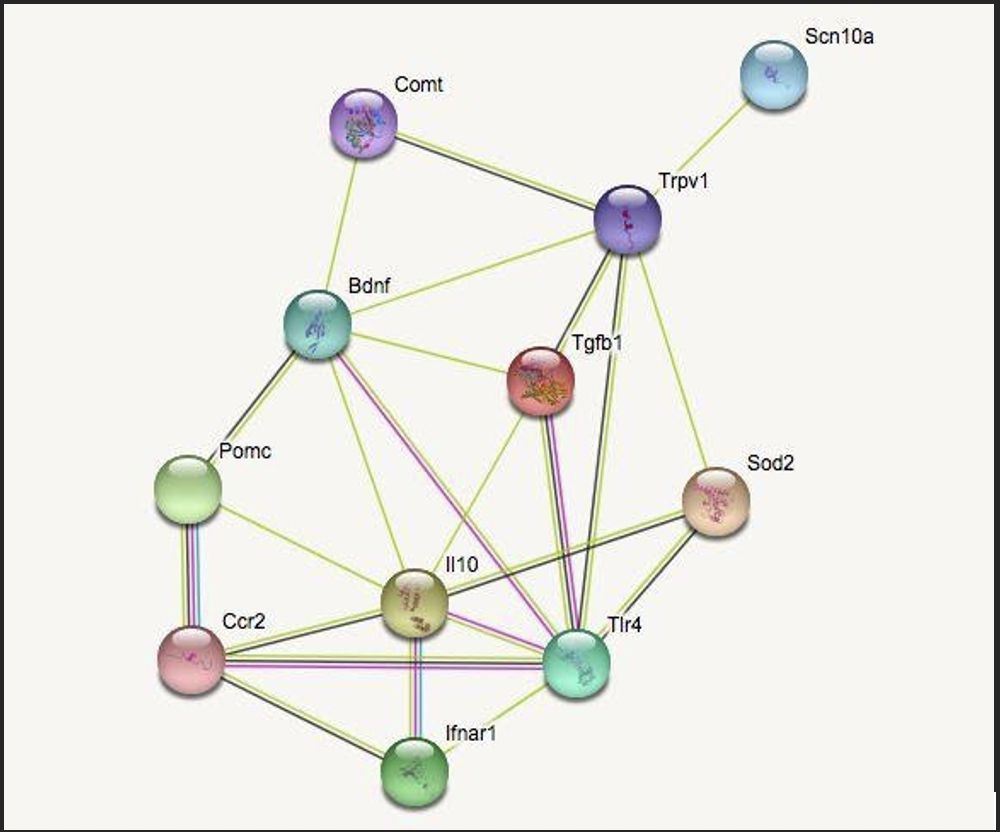
From our compiled list of 150 genes, the program identified 5 main clusters of proteins coded by the genes.
The functional clusters are genes which impact inflammation, neuropathic pain, cellular growth, and immune response. The fifth category, termed “other,” identifies genes which characteristically differ from the specified categories. Connections between two genes are indicated by a line drawn between the two. The two most correlated genes from each group (highest ranked by STRING) were chosen and mapped to see the quality and quantity of the cross-genomic interactions. The genes identified were BDNF & COMT (cellular growth), POMC & CCR2 (immune response), TRPV1 & SCN10A (neuropathic pain), IL10 & TLR4 (inflammation), and SOD2 & TGFB1 (other).
The selected genes were entered into STRING a second time to narrow the potential critical links between them. As seen in Figure 1, the correlative strength and quantity of connections varies. IL10, BDNF, and TRPV1 were the most connected genes shown. STRING shows that these genes are involved in the defense response of the human body, such as cytokine binding, with an abundance of genes being expressed in dendritic cells. Dendritic cells (which function as antigen presenting cells in the immune system) can induce a primary immune response in otherwise inactive T-lymphocytes and therefore play an important role in relaying information between the innate and adaptive immune system.
DISCUSSION
This project was conducted by entering pain-associated genes intThe goal of this study aimed to investigate the role of the neuropathic element in the larger scheme of an inflammatory response with the overarching goal of developing therapeutics for neuropathic pain in RA. Through this study, several genes were identified as potential targets for therapeutics.
In recent years, researchers have developed monoclonal antibodies, or laboratory-made antibodies aimed to bolster the immune system defense, directed against cytokines to reduce disease burden particularly in inflammation and pain. However, the treatment has not completely arrested long-term bone destruction in RA.⁶ Many of these monoclonal antibodies target individual cytokines. Additionally, our research did not identify the tumor necrosis factor (TNF) as a key variable in RA induced neuropathic pain, indicating that its correlative strength and quantity of connections did not merit a high enough ranking in STRING. TNF is the most commonly targeted cytokine by existing RA drug therapies, so it is unusual this gene would not show up in our meta-analysis. A possible explanation is that TNF is involved in nociceptive pain and inflammation but not neuropathic pain. The following are the highlighted target genes of this study.
Our search uncovered type I interferon receptor (IFNAR1) and IL10 as significant to RA. In standard immune responses, type I interferons have an important role in immune defense. However, RA patients demonstrate persistent activation of the type I interferon pathway. Overactivation of this pathway has been shown to increase inflammatory response.⁷ These results indicate that IL10 has anti-inflammatory and immunoregulatory roles that suggest a potential therapeutic role in RA. The Janus kinase (JAK) inhibitors reduce the downstream signaling pathways for many cytokine receptors including the type I interferon pathway and the IL10 receptor, thus regulating the pathways which cause inflammation. How- ever, these kinase inhibitors are only effective in roughly two-thirds of RA patients.⁷ Type I interferon (IFN) expression in the joints of arthritic mice is regulated by interferon regulatory factors (IRF) 3 and 7. These IRFs can be translocated to the nucleus by a variety of immune stimuli including toll-like receptor ligands (a class of pattern recognition proteins used to identify foreign molecules in the body), which induce an immune response if a virus is detected in the body.⁸ In the literature, IFNAR1 deficient mice exhibited osteopenia (weakening of the bones) with increased production of osteoclasts which degrade bone, indicating that IFN signaling may decrease osteoclast differentiation. Receptor activator of nuclear factor kappa-Β ligand (RANKL) maintains bone homeostasis through c-Fos-dependent induction of IFN-β.⁸ As a regulatory loop, IFN-β strongly inhibits the osteoclast differentiation by interfering with the RANKL-induced expression of c-Fos.⁹ This regulatory loop can be a target for future therapeutics and should be studied further. Osteoclasts have been implicated in sustaining the signals for neuropathic pain.
Additionally, several target genes identified in the search have been explored as possible targets for therapeutics in existing literature. Brain-derived neurotrophic factor (BDNF) is a neurotrophin with functions related to neuronal survival and proliferation processes as well as inflammation. BDNF is also an important central pain mediator. Severe RA patients reportedly express high levels of BDNF; however, BDNF expression has been shown to decrease in response to anti-TNF treatment.¹⁰ Another target is transforming growth factor (TGF) β1, which is expressed in the rheumatoid synovium. TGF-β1 contributes to the progression of inflammation and joint destruction in RA, an effect specific to arthritic synovial fibroblasts.⁵ Again, TGF-β1 can be a future target for therapeutic study and more research should be conducted on its mechanisms. Finally, transient receptor potential vanilloid subtype 1 (TRPV1) is best known for its function in nociception, especially in response to heat and inflammatory compounds. The literature suggests that it serves a similar function in joint afferent neurons, which encode information of joint movement, position, and nociception. Animals with Trpv1-/- exhibit lower levels of pain. When present, TRPV1 may therefore play a role in facilitating the joint damage and swelling that is characteristic of arthritis and therefore should be a target for future therapeutic study.¹¹
CONCLUSIONS
This project was conducted by entering pain-associated genes into STRING to demonstrate that inflammatory genes are highly connected and involved in numerous biological pathways, therefore implicating multiple specific inflammatory mechanisms that may be associated with neuropathic pain. The findings from the virtual model also elucidated several new target genes of interest for further study. This is especially important for the development of new, effective, and specialized strategies for RA pain therapeutics. Moving forward, we would like to study the role of these new target genes in RA inflammatory pathways as well as acute and chronic pain in mouse models in order to further elucidate the relationship between tactile pain and paw swelling. Finally, we would also like to explore gender differences between mice with these genetic deficiencies to better understand the gender disparity in RA diagnoses.
ACKNOWLEDGEMENTS
Thank you to Dr. Maripat Corr and lab members Peter Pham, Gwendallyn Stilson, and Valerie Hsu, Dr. Yaksh and his lab members, and the Undergraduate Research Scholarship Ledell family research program.
REFERENCES
- Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6:15.
- Elliott MJ, Maini RN, Feldmann M, et al. Repeated therapy with monoclonal antibody to tumour necrosis factor alpha (cA2) in patients with rheumatoid arthritis. Lancet. 1994;344(8930):1125–1127.
- Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;17:78.
- O’Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis 2013;72 Suppl 2:ii111–5.
- Sweeney S.E., Kimbler T.B., Firestein G.S. Synoviocyte innate immune responses: II. Pivotal role of IFN regulatory factor 3. J. Immunol. 2010;184:7162–7168.
- Troutman, Ty Dale et al. “Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K.” Cell cycle (Georgetown, Tex.) vol. 11,19 (2012): 3559-67.
- Takayanagi, H., S. Kim, K. Matsuo, H. Suzuki, T. Suzuki, K. Sato, T. Yokochi, H. Oda, K. Nakamura, N. Ida, et al. 2002. RANKL maintains bone homeostasis through c-Fos–dependent induction of interferon-β. Nature. 416:744–749.
- Takayanagi, H., Kim, S., & Taniguchi, T. (2002). Signaling crosstalk between RANKL and interferons in osteoclast differentiation. Arthritis research, 4 Suppl 3( Suppl 3), S227–S232.
- Kouskoff V, Signorelli K, Benoist C, Mathis D. Cassette vectors directing expression of T cell receptor genes in transgenic mice. J. Immunol. Methods. 1995;180:273–280.
- Forsgren, Sture et al. “Measurements in the Blood of BDNF for RA Patients and in Response to Anti-TNF Treatment Help Us to Clarify the Magnitude of Centrally Related Pain and to Explain the Relief of This Pain upon Treatment.” International journal of inflammation vol. 2011 (2011): 650685.
- Galindo T., Reyna J., Weyer A. (2018). Evidence for transient receptor potential (TRP) channel contribution to arthritis pain and pathogenesis. Pharmaceuticals 11:105. 10.3390/ph11040105
Written by Asim Mohiuddin
Neurobiology Major, Eleanor Roosevelt. PI: Dr. Maripat Corr, UC San Diego Department of Rheumatology, Allergy & Immunology