
A marine microbe Salinispora tropica produces the salinosporamide A proteasome inhibitor that exhibits potent anti-cancer activity targeted particularly against glioblastoma. The Moore Lab at UC San Diego currently researches salinosporamide A, opening up the potential of future proteasome inhibiting drugs.
Deep in the Caribbean Sea, a small marine microbe known as Salinispora tropica coexists with its environment, relying
on intracellular chemical communication to interact with neighboring microorganisms. Over centuries of evolution, this microbe has become reliant on certain specialized metabolites used for ecological interaction rather than organismal growth to potentially defend against microbial predators by inhibiting certain cellular components, as well as maintain homeostasis. Surprisingly, diseased cancer cells are reliant on similar cellular components to rapidly proliferate, and thus these microbial metabolites have the potential to inhibit these proliferative mechanisms.
According to Dr. Bradley Moore of Scripps Institute of Oceanography and Skaggs School of Pharmacy, marine microbes serve as a promising source of biogenic molecules with varying biomedical applications, bolstered by the vast diversity and ingenious evolutionary mechanisms among the numerous marine microbiomes in the ocean. In particular, Dr. Moore has been exploring S. tropica’s unique metabolic products to elucidate whether these compounds may serve as potential treatments against glioblastoma, a particularly dangerous form of brain cancer.
Current Glioblastoma Treatments
Glioblastoma is the most common type of brain tumor, and accounts for almost half of all brain cancer cases. Glioblastoma forms in glial cells which are cells in the central nervous system (CNS) that support and protect neuronal structure, function, and wellbeing. Specifically, an extremely abundant form of glial cells known as astrocytes are essential in maintaining the proper function of neuronal synaptic connections, thus sustaining proper neuronal communication in the brain. Glioblastoma, in turn, forms from cancerous astrocyte cell groups and moves aggressively to infiltrate healthy astrocytes surrounding the tumors, disrupting the astrocytes’ role in synaptic connection and impairing cognitive function. As in all forms of cancer, dysregulation of the normal cell cycle prevents the cycle from ending, causing excessive astrocytic growth in the form of glioblastoma.¹
Anticancer drug therapies are promising options to treat various types of cancers, and effective chemotherapies remove tumors where surgical removal is not feasible. Glioblastoma cannot be removed surgically because the processes of cancerous astrocytes surround the endothelial cells of the blood brain barrier and branch out with their long tentacle-like extensions, making the glioblastoma located near the delicate vasculature difficult to remove. With a median patient survival time without treatment of six months from onset, and a survival rate of only 40% in the first year after diagnosis, alternative treatment options are clearly needed to address this illness.
However, treating brain tumors involves a complicating factor: the blood-brain barrier (BBB). Analogous to a security screener, the BBB is impermeable to large polar compounds, while selectively allowing lipid-based, water insoluble compounds to enter the brain. This feature protects the brain from harmful toxins potentially circulating in the bloodstream and vitally controls the compounds that can enter the brain from the bloodstream.²
However, the BBB’s selectivity prevents most anticancer drugs from entering the CNS due to their polarity and large size, which present a serious difficulty in treating glioblastoma with pharmaceuticals. With many current candidate anticancer drugs unable to pass through the barrier, there remains a need for drugs with disparate structures. Interestingly, the Moore Lab has found that such a biochemical solution may be within the deep-sea bacteria S. tropica.
The Unusual Structure of Salinosporamide A
S. tropica belongs to the genus Salinispora discovered off the Bahamas by UC San Diego Professors William Fenical and Paul Jensen, who also pioneered the discovery of salinosporamide A compound. This microbe is part of a marine bacterium family called actinomycetes. These filament-shaped microbes contain specialized metabolites that serve as appealing pharmaceutical candidates. These specialized metabolites are not essential for organismal growth and reproduction, and instead are critical to the bacterium’s environmental response mechanisms, such as adaptation and defense systems.³ After genomic sequencing of S. tropica, various chemical laboratory techniques revealed unique biochemical structures that fascinate the Moore group; however, the specialized metabolite toxin known as salinosporamide A poses particular promise to anticancer drug therapy research.
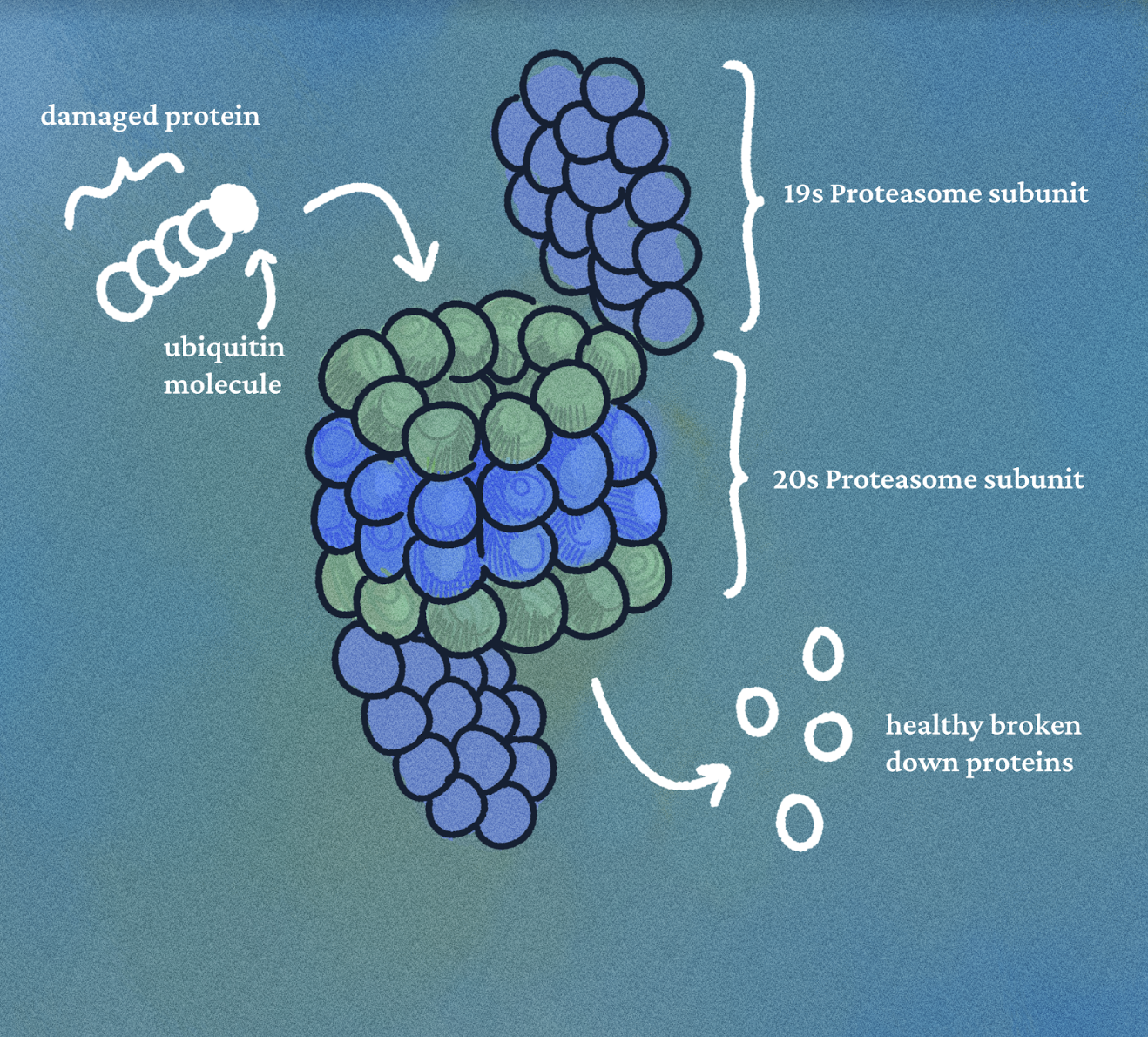
Salinosporamide A has potent proteasome inhibitory activity, causing a chain of chemical reactions that can ultimately
result in potent anticancer activity and use as an anticancer drug. The proteasome is a large protein complex that regulates protein deterioration, with the ability to promote apoptosis, or programmed cell death. When cellular proteins misfold or
become damaged, the enzyme ubiquitin ligase attaches a ubiquitin protein tag to the damaged protein, signaling for its degradation by the proteasome. Oftentimes, the damaged proteins are hydrolyzed into their amino acid building blocks which can be used to make new, healthy proteins. However, cancerous tumors often form large amounts of damaged proteins that are tagged and broken down in the proteasome, allowing them to be reused and contribute to effective cancerous growth. This results in greater proteasome activity in cancerous cells, and this greater activity is crucial to the cancer proliferative mechanism. When salinosporamide A binds to and inhibits proteasome 20S in astrocyte cells, protein recycling is blocked, allowing the cell to apoptose and thus preventing excessive cancerous growth with relatively minimal cytotoxicity in healthy cells. While this inhibitory regulation mechanism seems straightforward, the process is complex and relies on the unusual chemical structure of the salinosporamide A.
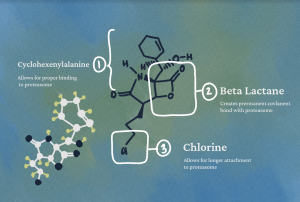
There are several biochemical characteristics of salinosporamide A that enable its proteasome regulation effects. The presence of cyclohexenylalanine, a six-membered carbon ring with amine and carboxylic acid groups, enables salinosporamide A to react with the 20S proteasome by positioning it for proper binding. Another vital structural component of salinosporamide A is a beta lactone, or a square-shaped heteroring composed of three carbons and an oxygen attached to an additional carbonyl group, an oxygen double-bonded to a carbon. This beta lactone has similar features to beta lactam rings, which are found in the structures of many antibiotics. A nucleophilic threonine residue hydroxyl in the proteasome attacks the electron deficient beta lactone carbonyl group, which after a cyclic rearrangement, creates a permanent bond between the proteasome and the lactone. Finally, a chlorine atom on one of the salinosporamide A side chains displaces to allow the beta lactone ring, after being opened by the threonine residue nucleophilic attack, to cyclicize, thus giving salinosporamide A the structural space to bind to the proteasome irreversibly. This stable bonding enables the metabolite to hold a longer residence time on the proteasome than other proteasome inhibitors, a quality crucial to its application in a pharmaceutical setting.
Unlike many other pharmaceutical molecules, salinosporamide A can permeate through the BBB by facilitated transport, or through selective channels spanning across the BBB. The Moore Lab determined that the structure of salinosporamide A mimics that of an amino acid due to its small, cyclic, and folded protein-like features. As such, it is classified as a nonproteinogenic amino acid since it contains an amine group and a carboxyl group connected to an organic carbon chain. This nonproteinogenic distinction comes from it being structurally distinct from the twenty-two amino acids created naturally from the genome. Amino acids can be transported across the BBB by facilitated transport, and thus salinosporamide A’s structural similarity to endogenous amino acids permits it to disguise itself as an amino acid and enter the CNS.
However, how does S. tropica create salinosporamide A without damaging its own proteasome? Normally, S. tropica produces both the proteasome inhibitor salinosporamide A and two copies of a proteasome during its natural metabolic processes. While this may seem counterintuitive, the bacterium evolved to introduce a mutation in the PSMB5 gene that allows one of the two co-synthesized proteasomes to retain functionality despite the existence of the toxin salinosporamide A. Interestingly, this fact may also shine light on ways cancer cells might resist treatment with salinosporamide A.
As more drugs are created to treat various diseases, evolutionary pressure drives biological components targeted by these drugs to evolve mutations that resist pharmaceutical treatments, resulting in resistance to these drugs accumulating over time. Proteasome resistance appeared in patients taking Bortezomib, the first proteasome inhibiting drug approved by the Food and Drug Administration for treatment of multiple myeloma. Although poorly understood, the resistance of this drug involves mutation in the gene PSMB5, coding for the subunit beta 5 of the 20S proteasome, which is partially responsible for breaking down proteins. Surprisingly, these mutations are similar to those in S. tropica, and occurs in the same location on the PSMB5 gene as the acquired resistance to Bortezomib in humans, suggesting a similar association between the drug resistance in humans and the evolutionary resistance in S. tropicas’ redundant proteasome.â· With further scientific exploration into how the secondary proteasome interacts with salinosporamide A and functions within the greater S. tropica proteasome complex, the mechanics of resistance to proteasome inhibiting drugs may become better understood in order to combat the growing health concern.
Synthesizing Salinosporamide
The many unique characteristics of salinosporamide A make it a viable pharmaceutical inhibitor qualified for further research. The Moore Lab now organically synthesizes the molecule, as opposed to deriving it from the S. tropica organisms’ natural metabolic processes. Now that mass production is viable, the compound has been clinically denoted as Marizomib, an oral anticancer drug currently in stage three clinical trials at UC San Diego for treatment against glioblastoma.
The Moore Lab is currently exploring how the chemical structure is formed biologically, specifically the unique beta lactone ring. Kate Bauman, a graduate student heading the project on salinosporamide A in the Moore Lab, recently identified a novel enzyme that S. tropica utilizes to form the beta lactone, as well as a gamma lactam, or a pentagon ring structure. While necessary but not directly responsible for the inhibitory properties of the toxin mentioned earlier, the gamma lactam is particularly difficult to artificially synthesize, making the enzyme’s production of it especially notable.
This enzyme is responsible for the final chemical assembly of salinosporamide A, and the use of this enzyme in the lab may replace the need to synthesize the molecule through artificial organic methods. The structure of the beta lactone and gamma lactam require a complicated and expensive synthesis process that produces small yields of Marizomib, creating limited access for salinosporamide A production. Using the natural enzyme to form this compound will cut down the economic and environmental costs of its production, allowing higher yields and accessibility to the compound.
Additionally, the ability to synthesize beta lactone and gamma lactam will enable the creation of a whole class of salinosporamides, each with their own slight variation in chemical structure. Specifically, a salinosporamide that is not naturally made could be tailored to inhibit the proteasomes of particular diseases with increased proteasome activity, for example non-glioblastoma cancers. While the impact of this enzyme discovery on Marizomib is yet to be revealed, there certainly is a multitude of potential applications in utilizing the drug to achieve proteasome inhibition.
With this major breakthrough, Dr. Moore is hopeful that glioblastoma treatment could be just one application for salinosporamide molecules. The potential of proteasome inhibition extends to the multiple types of proteasomes other than the 20S protease involved in glioblastoma; other potential targets include immunoproteasomes, which are associated with neurological diseases such as Huntington’s Disease. Additionally, infectious diseases such as tuberculosis could be affected by proteasome misregulation. Clearly, further research is essential to revealing the numerous applications of proteasome inhibition in current medicine.
The wide possibilities of proteasome inhibitor treatments provide a glance into the potential of natural product research. If just one toxin from a tiny microbe living at the bottom of the ocean can provide such promising treatment against brain cancer, imagine how many other organisms hold the secret to curing diseases within their unique molecular structure and fascinating biosignaling pathways. With time, and additional scientific innovation, exploration, and research, perhaps we will become accustomed to searching for solutions to humanity’s most daunting health challenges in the hidden science of
the natural world.
References
- Wirsching HG, Galanis E, Weller M. 2016. Glioblastoma. 134:381-97.
- Mikitsh JL, Chacko AM. 2014. Pathways for Small Molecule Delivery to the Central Nervous System Across the Blood-Brain Barrier. Perspectives in Medicinal Chemistry. 6:11-24.
- Jensen PR, Moore BS, Fenical W. 2015. The Marine Actinomycete Genus Salinispora: A Model Organism for Secondary Metabolite Discovery. Natural Product Report. 32(5):738-751.
- Adams J. 2003. The proteasome: structure, function, and role in the cell. Cancer Treatment Reviews. 29(1):3-9.
- Kale AJ, Moore BS. 2012. Molecular Mechanisms of Acquired Proteasome Inhibitor Resistance. Journal of Medicinal Chemistry. 55:10317-10327.
- Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. 2003. Salinosporamide A: A Highly Cytotoxic Proteasome Inhibitor from a Novel Microbial Source, a Marine Bacterium of the New Genus Salinospora. 2003. Angewandte Chemie International Edition. 42(3):355-357.
- Niewerth D, JAnsen G, Riethoff LFV, Meerloo JV, Kale AJ, Moore BS, Assaraf YG, Anderl JL, Zweegman S, Kaspers GJL, Cloos J. 2014. Antileukemic activity and mechanism of drug resistance to the marine Salinispora tropica proteasome inhibitor salinosporamide A (Marizomib). Molecular Pharmacology. 86(1):12-9.
Written by Emily White
Emily is a Molecular and Cell Biology/Public Health Major from Seventh College. She will be graduating in 2024.